
Post Electrophoretic Analysis Articles
Heteroduplex Analysis
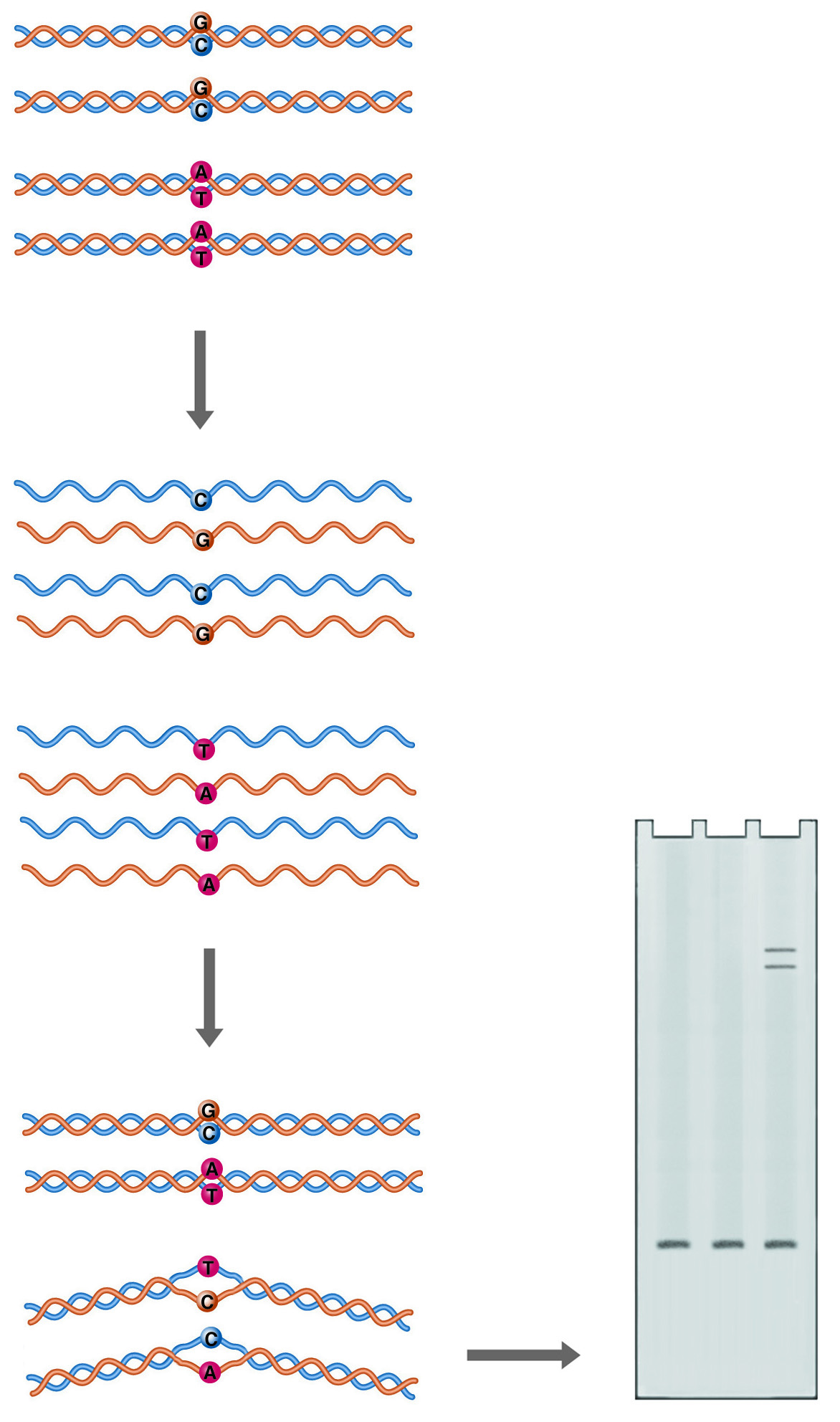
Heteroduplex Analysis. Annealing of mutant DNA to wild-type probe gives duplexes with one or more mismatched bases (heteroduplexes). Mismatching causes the double helix to take on a conformation that retards its mobility during electrophoresis.
Double-stranded DNA is not a completely straight rigid rod. Sequence variations can cause bends in the double helix, or even alter the basic structure of the helix. A bend or kink in the DNA restricts its mobility through a sieving matrix, as the bent molecule presents a larger projected area to the gel pores. A mismatch between the two strands of DNA in a duplex can produce a more radical kink in the structure, producing a heteroduplex species which can easily be resolved from the homoduplex by electrophoresis.
In this system, control DNA is denatured and allowed to anneal with denatured sample DNA. The renatured products are analyzed on a gel optimized to resolve conformational differences, such as National Diagnostics' SequaGel MD. If the sample DNA is not identical to the control DNA, multiple bands are observed. The fastest migrating band is the homoduplex control and/or the homoduplex sample. Heteroduplexes with mismatches migrate more slowly.
Heteroduplex analysis is easily applied to large numbers of samples and is particularly suited to the analysis of PCR products because both sample and control DNA must be of the same size. In PCR, this would correspond to using the same amplification primers.
Heteroduplex Analysis Guideline
GEL PREPARATION (0.8 to 1.0 mm thick):
- Preparation of Working Solutions:
Combine 50 mL SequaGel MD, 6 mL 10X TBE and (optionally) 15 g urea in an Erlenmeyer flask. Fill to 100 ml with deionized water and mix thoroughly. Urea may assist the formation of more distinct bands during electrophoresis and may reduce the formation of doublets in homoduplex controls. - Casting the gel:
Add 40 µl TEMED and 400µl freshly prepared 10% ammonium sulfate to the solution. Treat one plate from the gel cassette with Glass Free to facilitate later disassembly of the cassette. Using the standard procedure, pour the gel solution into the cassette, insert the comb, and allow to polymerize at room temperature for a minimum of 60 minutes. Attach the gel cassette to the electrophoresis apparatus, and fill the upper and lower chambers with 0.6X TBE.
SAMPLE PREPARATION:
- PCR amplification:PCR conditions should be optimized for the desired PCR product before Heteroduplex Analysis. It is recommended that the minimum number of PCR cycles be used on a purified, salt free template, and that reagent and primer concentrations be optimized.After PCR thermal cycling, add EDTA to a final concentration of 5 mM (1µl of 0.5 M EDTA per 100 µl reaction) to inactivate the Taq DNA Polymerase.
- Hybridization:Mix equivalent quantities of wild type and sample PCR-amplified DNA. Heat at 95°C for 3 minutes. Afterward, cool the mixture to room temperature during a 20-30 minute time period. A thermocycler can help facilitate this step.
ELECTROPHORESIS:
- Add 1 µl Triple Dye Loading Buffer (provided in kit) per 5 µl of sample and mix thoroughly.
- Rinse the wells with running buffer and load the samples in the 1.0X SequaGel MD gel. One lane should consist of control homoduplex DNA, and one of the sample homoduplex DNA. This will allow the detection of non-heteroduplex artifacts on the gel. Another lane should consist of an appropriate DNA size marker.
- Run the gel in 0.6X TBE, at a constant voltage of 20 V/cm, as determined by the length of the gel. For a 40 cm gel, set the power supply to 860 V. Approximate run times can be estimated from the chart below:
| Fragment Size (bp) | Run Time (hours @ 800V) | Volt X Hours |
|---|---|---|
| 200 | 14.0 | 11,200 |
| 250 | 14.5 | 11,600 |
| 300 | 16.5 | 13,200 |
| 500 | 20 | 16,000 |
| 700 | 25 | 20,000 |
| 900 | 30 | 24,000 |
When the electrophoretic run is complete, remove the gel from the apparatus, and carefully remove one plate from the gel. Stain with ethidium bromide or silver stain.
STAINING:
Stain using 0.6X TBE containing 1 mg /ml of ethidium bromide. Water should not be used in place of the TBE, because the gel will swell when placed in water. Stain for 15-30 minutes. For maximum sensitivity, destaining in 0.6X TBE for up to 30 minutes may be required.
To visualize ethidium bromide-stained bands, cover the gel with plastic wrap and place the plate with the gel side down on a UV-transilluminator. It may assist in handling and visualization to cut out the gel region containing the bands of interest.
WARNING: ETHIDIUM BROMIDE IS BELIEVED TO BE A CARCINOGEN AND SHOULD BE DISPOSED OF PROPERLY.
Silver staining may be used to increase band visibility.
NEXT TOPIC: SSCP Analysis