
Post Electrophoretic Analysis Articles
Sanger Sequencing
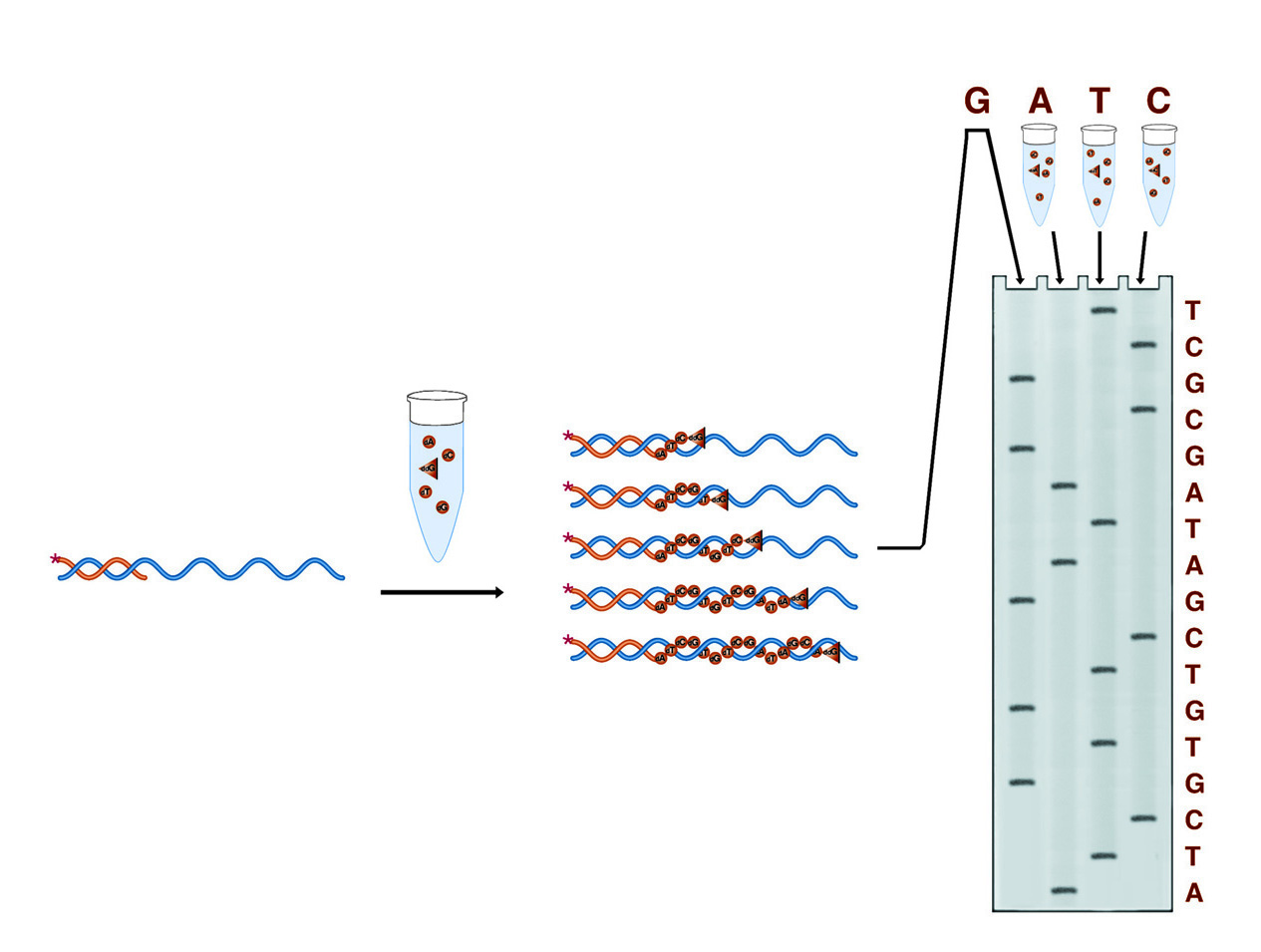
In Sanger sequencing four reactions are run, each designed to terminate the growing DNA chain at one of the four bases (the G reaction is shown in detail). The result is four collections of fragments whose comparative lengths indicate the positions of the four bases (the sequence) of the DNA under study.
In Sanger dideoxy terminator sequencing, the sample DNA is used as a template for a DNA polymerase. Four polymerase reactions are carried out involving enzyme, primer, and sample DNA, along with dNTPs. Each reaction also contains one of the four dideoxy NTPs. When a dideoxy NTP is added, chain lengthening terminates because ddNTP nucleotides lack 3' hydroxyl groups by which to form the next phosphodiester bond. Each reaction contains one of the four bases as a dideoxy NTP, thus each reaction results in fragments terminating at that base. The four reactions produce four collections of fragments with lengths reflecting the sequence positions of each of the four respective bases.
Numerous commercial kits are available for Sanger sequencing. These kits provide excellent and consistent results without the need for the researcher to titrate dideoxy mixtures for maximum efficiency. An outline of the method is presented here. However, it is strongly recommended you follow the protocol provided with your particular kit.
For optimum performance, dideoxy sequencing requires a single-stranded substrate. The most prevalent artifact associated with dideoxy sequencing is the appearance of BAFLs (Bands Across Four Lanes), which are due to polymerase "pause" sites, at which the enzyme tends to fall off of the DNA, resulting in a non-dideoxy termination. This effect is exaggerated when the polymerase is required to process double-stranded DNA. Single-stranded substrates are generally prepared by cloning the DNA of interest into vectors derived from the bacteriophage M-13. Phage M-13 replicates its DNA in the bacterium in double-stranded form, allowing easy cloning manipulations, but its phage form contains circular single-stranded DNA.
Purifying SS M-13 DNA
- Precipitate the phage with Polyethylene Glycol (PEG):
- Remove bacteria from 1.5 ml of culture by centrifuging at 12 - 14 K RPM in a microcentrifuge.
- Transfer the supernatant to a fresh tube, containing 0.2 ml 2.5M NaCl + 20% PEG 8000 and mix well. Incubate for 15 minutes at room temperature.
- Pellet the phage particles in a microcentrifuge at 14K RPM for 15 minutes at 4°C.
- Remove supernatant with a pipette, being careful not to disturb the very small phage pellet.
- Briefly centrifuge the tube to bring residual supernatant to the bottom and remove it with a pipette.
- Purify the DNA:
- Vortex the pellet in 50µl T10E1 buffer.
- Add 50µl phenol equilibrated with tris pH 8.0.
- Vortex 30 seconds - 1 minute.
- Separate the phases in a microcentrifuge at 14K RPM for 2 minutes.
- Recover the upper phase, being careful not to disturb the interface layer. (Note: leaving some of the supernatants over the phenol phase ensures a cleaner preparation.)
- Add 300µl ethanol and 15µl 3M sodium acetate, pH 5.2. Mix well and incubate at room temperature for 15 minutes.
- Collect the DNA in a microcentrifuge at 14K RPM for 20 minutes at 4°C.
- Remove the supernatant and wash the pellet twice with 70% ethanol.
- Proceed with primer annealing: 1.5 ml of culture should yield > 5µg of DNA, sufficient for 1-2 sequencing reactions.
Dideoxy Sequencing (Tag Polymerase)
- Anneal the primer:
Dissolve 0.5pmole of M-13 substrate in 10µl of Taq buffer: 50mM tris HCl, pH 9.0, 15mM MgCl2. Add 0.5pmole of primer in 1µl of water or TE buffer. Incubate at 65°C for 10 minutes, then allow to cool to room temperature over 30 minutes. This is most easily accomplished by placing the tube in a 100ml beaker of water at 65°C and allowing the beaker to cool on the benchtop. - Set up the termination reactions:
Aliquot 2.5µl of A, C, G, and T termination mixtures into appropriately labeled tubes. The termination mixtures contain 8µm of the dNTP targeted for termination, 800µM each of the other 3 dNTPs, and 0.1mM EDTA. In addition, each mixture contains one ddNTP, as follows:
A: 125µM ddATP
C: 50µM ddCTP
G: 20µM ddGTP
T: 100µM ddTTP - Labeling reaction (initial primer extension):
Add 2µl of a mixture of 1.6µM each of all 4 dNTPs, 1µl 10mCi/ml 35S-dATP, and 2 units of Taq polymerase (diluted in Taq buffer to 2U/µl). Place in a 40-45°C water bath for 3-5 minutes. Do not start the reaction at 45°- this may denature some primer-template complexes. - Termination:
Add 3.5µl of labeling reaction to each termination tube. Mix well and incubate at 60°C for 5-10 minutes. Add 4µl of loading dye (95% formamide, 20mM EDTA, 0.05% each bromophenol blue and xylene cyanol), and heat to 95°C for 2 minutes prior to loading on a denaturing PAGE gel.
NEXT TOPIC: Pouring Sequencing Gels
- Using PAGE to Determine Nucleic Acid Molecular Weight
- SSCP Analysis
- Sanger Sequencing
- Sample Preparation for Native PAGE of DNA
- Sample Prep for Denaturing PAGE of DNA
- S1 Mapping
- Run Conditions in Denaturing PAGE
- RNA Mapping
- RNA Electrophoresis
- Ribonuclease Protection
- Restriction Digest Mapping
- Primer Extension
- Preparing Denaturing DNA & RNA Gels
- Preparation of Denaturing Agarose Gels
- Preparation of Agarose Gels
- Pouring Sequencing Gels
- PFGE and FIGE
- PCR Analysis: Yield and Kinetics
- PCR Analysis: An Examination
- Native PAGE of DNA
- Mobility Shift Assay
- Methylation & Uracil Interference Assays
- Maxam & Gilbert Sequencing
- Manual Sequencing
- In Gel Enzyme Reactions
- Heteroduplex Analysis
- Gel Preparation for Native PAGE of DNA
- Gel Electrophoresis of PCR Products
- DNase I Footprinting
- DNA/RNA Purification from PAGE Gels
- DNA/RNA Purification from Agarose Gels – Electroelution
- Differential Display
- Denaturing Polyacrylamide Gel Electrophoresis of DNA & RNA
- Conformational Analysis
- Automated Sequencers
- Analysis of DNA/Protein Interactions
- Agarose Gel Electrophoresis of DNA and RNA – Uses and Variations
- Agarose Gel Electrophoresis of DNA and RNA – An Introduction