
Post Electrophoretic Analysis Articles
Primer Extension
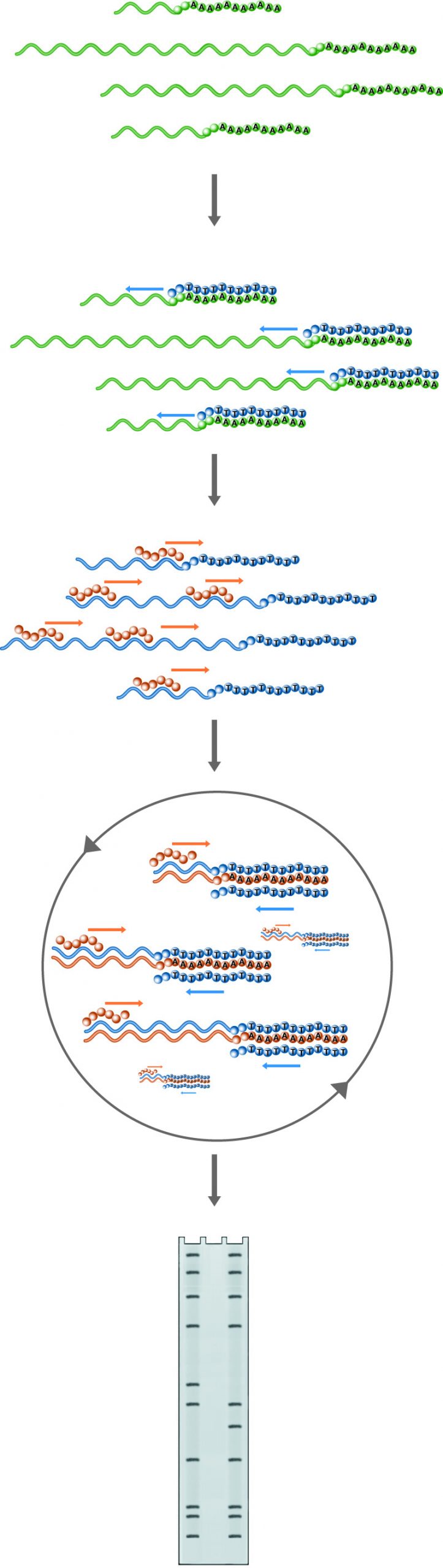
n Primer Extension, the probe introduced to the mRNA pool will hybridize with the RNA of interest if it is present. Hybrids are then extended by reverse transcriptase. The information gained through this method includes the confirmation of the presence of the RNA of interest, the location of the transcription start site, and if an excess of primer is used, the amount of the particular RNA present in the sample. Lanes: 1) Experimental, 2) Probe control, 3) Sequence ladder.
Primer extension is another technique used to analyze RNA structure and expression. In this method, an oligonucleotide primer is annealed to RNA and extended to a cDNA copy by reverse transcriptase in the presence of labeled dNTPs. Alternately, the primer is labeled and no label is included in the extension reaction. If the RNA of interest is present, extended products will appear on a denaturing gel. Furthermore, the size of the extended product will indicate the position of the 5' end of the RNA, and, if an excess of primer is used, the amount of cDNA produced will reflect the amount of target RNA in the sample.
Primer extension provides the same type of information as S1 mapping. However, primer extension is unaffected by splice sites. In cases where only a genomic probe is available and an intervening splice site prevents S1 mapping of the start site, primer extension offers a useful alternative. Primer extension offers additional advantages over S-1 mapping. A genomic clone of the target RNA is not required; only 30 - 50 bases of sequence need to be known to generate the primer. Additionally, probe preparation is easier, because the primer is single-stranded. This means that no elaborate procedures are needed prior to labeling.
Primer Extension
- Primer Selection and Preparation:
- Select a priming site that is 30 - 50 bases long, containing no self-complementary sequences. The site should be within 150 bases of the transcriptional start site, as reverse transcriptase has a tendency to find pause/termination sites in larger transcripts.
- End-label the primer using 32P ATP and T4 polynucleotide kinase.
- Use the buffer and protocol recommended by the enzyme supplier for best results. Labeling of 100 µg of primer should incorporate 1-5x107 cpm, or 3x105 cpm/µg.
- Remove unincorporated label by 3 rounds of precipitation with 1 volume 4M ammonium acetate and 10 volumes of ethanol.
- Precipitate for 30 minutes @ -70°C, and redissolve in 30 µl water between precipitations.
- Hybridization:
- Add 0.1µg (3x104 CPM) of a labeled probe to 50 µg RNA sample in 100 µl.
- Add 0.1 volume 3M sodium acetate and 2.5 volumes ethanol, and precipitate for 30 minutes at room temperature.
- Pellet, remove supernatant and allow the pellet to air dry for 15 minutes. Overdrying will make redissolving the pellet difficult.
- Redissolve RNA/probe in 30 µl of hybridization buffer (3M NaCl, 0.4M HEPES [pH 7.6], 1 mM EDTA).
- Hybridize overnight at 30 - 50°C (optimize temperature to reduce background).
- Precipitate 30µl hybridization with 150µl 0.3M sodium acetate and 500µl of ethanol.
- Wash pellet with 70% ethanol containing 30mM sodium acetate pH 5.3.
- Remove supernatant and allow the pellet to air dry for 15 minutes.
- Primer Extension Reaction:
- Redissolve sample pellet in a mixture of 18 µl H2O, 2.6 µl 10X RT buffer, 3.5 µl 4mM dNTP's and 2 µl RNase inhibitor.
- Add 400 units of reverse transcriptase (AMV). Allow the reaction to proceed at 42°C for 1.5 hours.Stop the reaction with 1 µl 500 mM EDTA.
- Digest substrate RNA with 1µg (1 µl of 1mg/ml) RNase A to prevent gel distortions. Digest 1 hour at 37°C. Extract reactions with phenol and then ethanol precipitate.
- Redissolve in 5 µl of water, add denaturing loading buffer, and analyze 2-5 µl on a denaturing PAGE gel.
NEXT TOPIC: Analysis of DNA/Protein Interactions