
Gel Electrophoresis of Proteins
Isoelectric Focusing
Conventional electrophoresis techniques can separate up to 100 different proteins on one run. Typically, cell or tissue extracts contain thousands of proteins, most of which will not be resolved into single bands using a separation based on any one parameter, such as size or net charge. For any one size range, there is a high probability of more than one protein (out of thousands) falling into this range. Separation on the basis of two parameters, usually size and isoelectric point, lowers the probability that two proteins will overlap, and allows the resolution of thousands of protein species on one gel. Such two dimensional separations are carried out by running a one dimensional gel to separate by the first parameter, and then laying this gel or a lane cut from it, across the top of the second dimension gel, the first gel serving as the "sample" for the second. Typically, the second dimension gel is an SDS PAGE gel, because this gives a direct measurement of a protein's size. The first dimension usually employs isoelectric focusing (IEP) as the means of separation. IEF gives a measurement of the isoelectric point (pI) of the protein - the pH at which the protein sample will not migrate in an electric field, the protein's net charge being zero. A two dimensional gel can give size and pI information on thousands of proteins in one run.
Further Exploration on Isoelectric Focusing
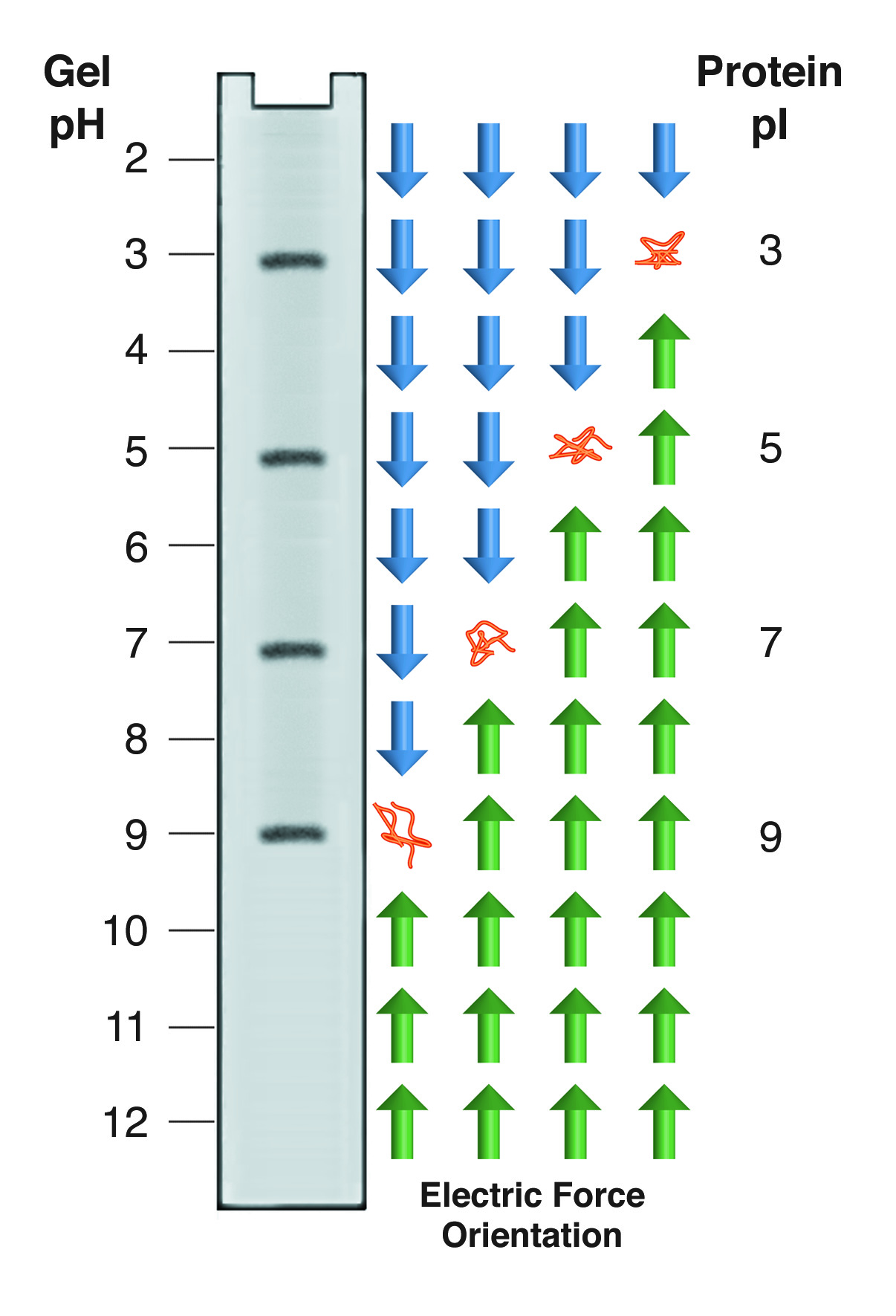
Isoelectric focusing employs a pH gradient extending the length of an electrophoresis gel. A protein stops migrating when it enters the zone in which the surrounding pH equals its isoelectric point, pI. At any other point in the gradient, the protein acquires a charge which causes it to migrate toward its pI (green and blue arrows).
Proteins carry charged groups on their surface. Each of these functional groups has a pK, which corresponds to the pH at which half of the members of that group are protonated. Above the pK that group can be considered fully protonated, below the pK, fully deprotonated. Thus, as the pH changes, the net charge on a protein's surface will change. At high pH, most proteins will have many deprotonated surface groups and will carry a net negative charge. At low pH, with many protons added to the surface, most proteins have a net positive charge. At some intermediate pH, different for every protein, the net charge on the protein will be zero. It is important to remember that this is a net charge - the protein is not uncharged - it carries equal numbers of positive and negative charges. The pH at which a protein has a net charge of zero is designated as its isoelectric point (pI).
A protein dissolved in buffer at its pI has no net charge and thus no net electrophoretic mobility. In isoelectric focusing (IEF) a pH gradient is established along the length of the gel. Proteins migrate through this gradient until they reach their pI. The gradient is set up so that negatively charged molecules migrate into a decreasing pH. If a protein is in a region where the pH is above its pI, it has a negative charge and moves to a lower pH. If it is in a pH below its pI, it has a positive charge which moves it into higher pH regions. This gives rise to the "focusing" aspect of IEF, as proteins are continually swept back into tight bands centered on the appropriate pI (see figure below). IEF is thus an equilibrium electrophoresis system, runs until protein movement ceases.
Ampholytes
The pH gradient in IEF gels is generated by the inclusion of ampholytes, low molecular weight amphoteric molecules. A mixture of ampholytes is used, each having a different pI. Like protein molecules, the ampholytes migrate through the gel until they reach a region where the pH is equal to their pI. Unlike the proteins, the ampholytes are present in high enough concentration to change their local pH. The gel is set up with a uniform mixture of ampholytes throughout, and its anodic and cathodic ends are immersed in dilute acid and base respectively. Ampholytes near the ends of the gels will be positively charged near the positive electrode, and negatively charged near the negative electrode. They, therefore, begin to migrate into the gel, with the most charged (ie the ones furthest from their pI) moving the fastest. Over time they separate into zones of defined pH. If the ampholyte system is well designed, a smooth gradient of pH is created, with no abrupt changes, or "steps." Commercial systems are available in a broad range (2 - 12 pH) or narrow range (extending 2 pH units across the gel).
Various mixtures of amphoteric substances have been used as ampholytes: amino acids, proteins, and poly acidic polybasic synthetic molecules. Natural amino acids have poor conductivity and poor buffering capacity in their zwitterionic state, making them poor candidates. Proteins can be good ampholytes, but they interfere with the analysis of the sample, by introducing new proteins into the mixture. Polycarboxylic acid polyamines are the most commonly used ampholytes. These molecules have excellent buffering capacity and conductivity across a broad pH range and are usually provided in a molecular weight range of 300 - 500, which is small enough to avoid interference with most subsequent processing. Their sole disadvantage is that they may bind tightly to the proteins, due to ionic interactions, and can be very difficult to remove.
Isoelectric Focusing
IEF is most frequently carried out as the first step in 2-dimensional electrophoresis. The apparatus best suited to this use of the gels is a "tube gel" system. The gels are cast and run in glass tubes with an internal diameter matched to the thickness of the second dimension gel. 1.5mm gels are commonly used. After the IEF gel has been run, it is extruded from the tube and laid across the top of the second dimension gel. This system is assumed in the protocol given below. IEF gels can also be run as slabs, which allows an increased sample throughput. Slab IEF gels can be cut into strips for loading onto second dimension gels, if desired.
-
- PREPARE THE GELS
- To formulate gel solution, dissolve 4g urea in 3 ml H2O + 1ml ProtoGel. Warm to 37°C if needed. De-gas for 10 minutes under aspiration.
- Add 150µl NP-40 detergent and 0.4ml ampholytes, mix and filter through a 0.22m filter.
- Add 30µl of 10% Ammonium Persulfate, and 3µl of TEMED. Mix well and place the solution in a 10ml syringe with a 22ga needle as long as the gel tubes.Insert the needle into the gel tube until it reaches the bottom, and fill the tube, withdrawing the needle as the fluid level rises. Use the needle to dislodge any bubbles. Allow the gels to polymerize for at least 2 hours.
- PREPARE THE GELS
-
- PREPARE THE SAMPLES
- To each gram of tissue, add 15ml of sample buffer, consisting of the following:
- 9M Urea
- 4% NP-40
- 2% Ampholytes (pH 9-11)
- 2% Mercaptoethanol pH to >9 with NaOH
- Homogenize if necessary. Incubate at room temperature for 10 minutes, then centrifuge for 1 hour at 100,000g. Remove the supernatant without disturbing the pellet, which contains materials likely to clog the IEF gel.
- To each gram of tissue, add 15ml of sample buffer, consisting of the following:
- PREPARE THE SAMPLES
-
- RUN CONDITIONS
- Fill the lower tank with 0.1% phosphoric acid. Place the gels in the apparatus and fill the upper tank with 20mM NaOH. Use a syringe to dislodge any bubbles from inside the gel tubes. Any bubbles in the tubes will distort the electric field and prevent gels from completely focusing during the run.
- Samples prepared as above can be loaded by layering onto the tops of the gels. Run at 500-700V (depending upon the apparatus used) for 16-24 hours. At the end of the run, mark the top end of each gel with a small amount of bromophenol blue.
- RUN CONDITIONS
-
- POST ELECTROPHORESIS
IEF gels may be coomassie stained, but most often they are loaded onto SDS PAGE gels for second dimension analysis. Extrude the gels by applying pressure to one end of the tube with a pipette or syringe, while holding the other end over a vial or tray. Extruded gels may be frozen at -70°C for later analysis.
- POST ELECTROPHORESIS
- LOADING AN IEF GEL ONTO A SECOND DIMENSION SLAB
- Place the IEF gel in a solution of 20% glycerol, 4% SDS, and 250mM Tris-HCl, pH 6.8, with 1% mercaptoethanol added just prior to use. Incubate the gel in this solution for 5-10 minutes.
- Dissolve 0.1g agarose in 20ml of Tris-Glycine-SDS buffer. Heat until the agarose is melted.
- Overlay the second dimension gel with 1-2 mm of agarose, place the first dimension gel over the agarose, being careful not to trap any air bubbles. Overlay the gel with another 1-2mm of agarose solution, and run the second dimension gel.
NEXT TOPIC: Visualizing Nucleic Acids - UV Shadowing